


Mol Ther Nucleic Acids | 临港实验室合作应用碱基编辑工具实现无义突变的精准修复及肌萎缩症治疗
杜氏肌营养不良( Duchenne Muscular Dystrophy, DMD)是一种致死性的肌肉遗传性疾病,是儿童最常见的肌营养不良。患病的孩子往往在4-5岁出现症状,在13岁前失去独立行走能力,20-30岁因心肺功能衰竭而死亡。目前该病暂无通用、特效的治疗方案,全球无数的科学家、医学家为了攻克该病坚持不懈地进行研究,而基因编辑修复正是其中新兴的治疗方向之一。
2024年3月7日,临港实验室胥春龙团队联合辉大基因杨辉/李国玲团队及福建医科大学附属第一医院陈万金/王柠团队在Molecular Therapy Nucleic Acids在线发表了题为“Correction of human non-sense mutation via adenine base editing for Duchenne muscular dystrophy treatment in mouse”的研究论文。该研究应用DNA单碱基编辑工具,在人源化的模型鼠中实现了无义突变的精准修复,为DMD的基因编辑治疗提供了新的证据支持。

现有的基因治疗方案,无论是外显子跳跃,还是通过mini-dsytrophin进行的基因增补,都是利用截短的dystrophin蛋白仍残留有部分功能这一机制,将症状较为严重的DMD转变为较轻的Becker型肌营养不良(Becker muscular dystrophy, BMD),从而减轻疾病表型,延长患者寿命,并没有在基因水平完全修复dystrophin基因。而在这项研究中,研究者运用单碱基编辑技术将无义突变DMD在DNA水平进行精确修复,完全修复了dystrophin基因。该研究首先在福建医科大学附属第一医院前期建立的DMD随访数据库的基础上,采用报告载体的方式对突变无义位点进行编辑效率筛选,发现接近一半位点在体外可被较高水平修复,随后在患者来源的iPSCs中验证了编辑效率及功能恢复情况,同时运用胚胎注射技术构建了c.2977C>T, p.Gln993*及c.4174C>T, p.Gln1392* 两种含有无义突变序列的人源化DMD小鼠模型,最后使用AAV9包装split-SpG-ABE系统进行成体基因治疗,在成体中证实了该系统的修复效果。
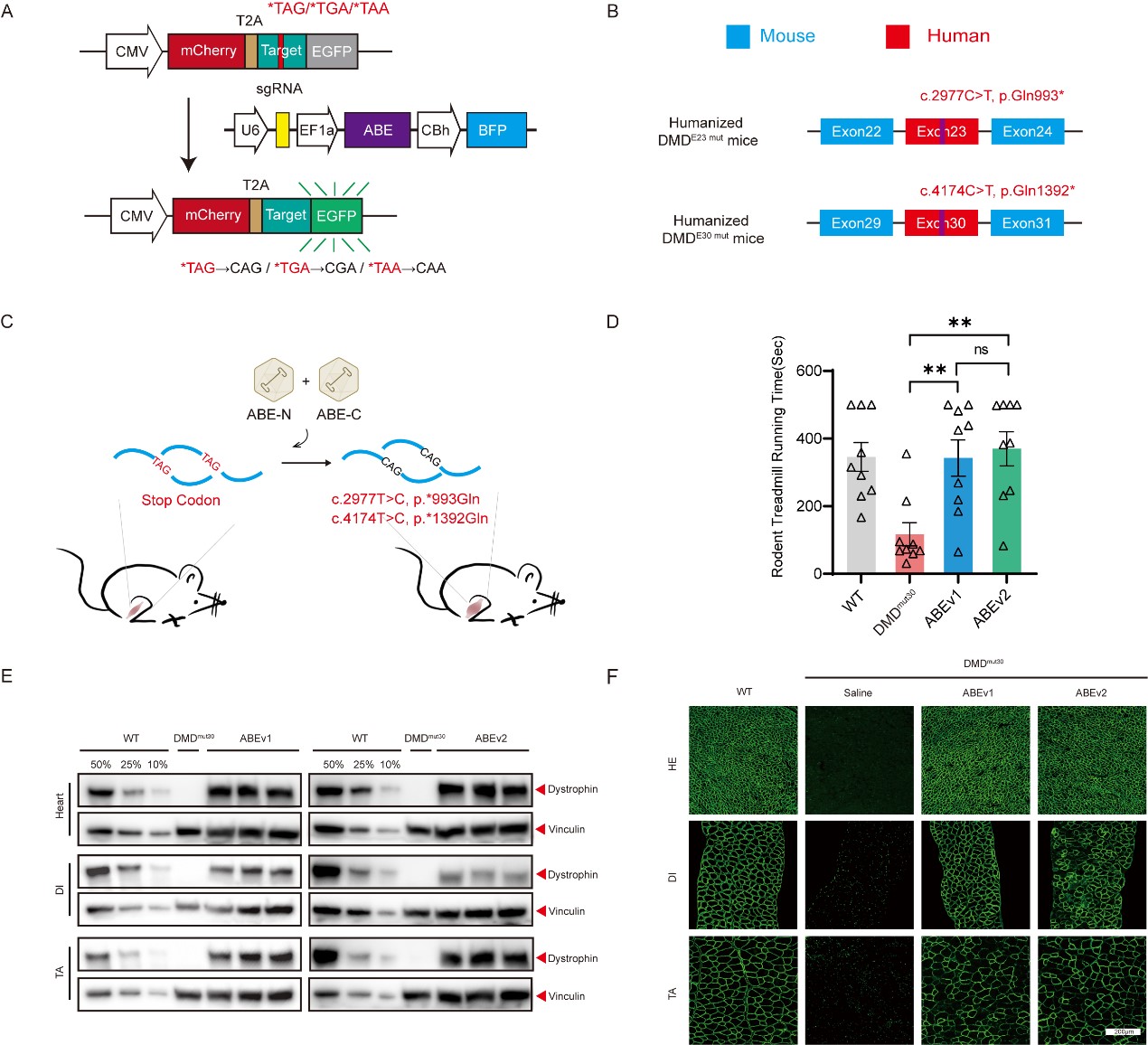
图1. 无义突变DMD的基因修复研究
A:无义突变报告载体sgRNA筛选方案模式图;B-C:人源化DMD无义突变模型鼠构建方案及治疗策略;D:全身给药治疗后运动功能恢复情况(小动物跑步机);E:全身给药治疗后心脏、膈肌及胫前肌dystrophin蛋白WB;F:全身给药治疗后心脏、膈肌及胫前肌dystrophin蛋白免疫荧光染色。
福建医科大学附属第一医院的金铭博士、林佳佳博士生,辉大基因李海森博士以及临港实验室李智方博士为该论文共同第一作者。临港实验室胥春龙研究员、辉大基因的李国玲博士,福建医科大学附属第一医院的陈万金教授及中科院脑智卓越中心杨辉研究员为共同通讯作者。相关工作得到国家自然科学基金委、科技创新2030-“脑科学与类脑研究”重大项目,福建省卫健委重大科研项目、临港实验室、上海科委启明星计划(A类)等资助。
附件下载: